Perl for Biologists
A collection of episodes with videos, codes, and exercises for learning the basics of the Perl programming language through genomics examples.
Greetings! Thank you for joining us.
Here, you will find a series of episodes featuring examples from genomics to help you learn the basics of the Perl programming language. No prior programming experience or knowledge of genetics is required.
Each episode includes a video and a working code highlighting a particular aspect of Perl in the context of a genomics problem. The progression from episode to episode is nearly linear. After completing the final episode, you will be able to download a genome file from NCBI, and search and tally intricate motifs.
Perl for Biologists arose out of a bold initiative launched ten years ago at the University of Miami to increase the number of physician-scientists. The innovative undergraduate PRISM (Program for Integrated Science and Mathematics) program includes a semester-long course in computing. The material herein constitutes about one-third of this course - Computing for Scientists.
Mastering a programming language is best accomplished through active participation. As you watch each video, please code along with us, pausing as needed. It is critical to complete all exercises, as they enable you to assess your progress towards becoming an independent computational scientist.
In closing, we would like to express our gratitude to those who have contributed to both the conception and the realization of Perl for Biologists. Colleagues Dimitris Papamichail and Burt Rosenberg, as well as former graduate assistants Andreas Seekircher and Justin Stoecker, helped with the development and testing of earlier versions of this material. Through their enthusiastic participation, our students aided us in fixing our ideas and setting realistic bounds for our own enthusiasm. In the final stages of the project, we benefitted from the assistance of Ziya Arnavut, Brian Coomes, Jason Glick, Denizen Kocak, Nancy Lawther, Joseph Masterjohn, Kenneth Palmer, Odelia Schwartz, and Stefan Wuchty.
Computational science is a flourishing frontier. It is our hope that Perl for Biologists episodes will allow the subject of computational biology to come alive and tempt you to explore it further.
Hüseyin Koçak and Basar Koc
Department of Computer Science
University of Miami
Introduction to Perl
Hello Perl
Download

Scalars
Download

Buggy
Download

#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: print.pl # # print collects strings in double quotes separated by commas # until the end of statement character ; # print function while outputting text enclosed in double quotes # replaces the variable names with their values (interpolation). # print does not interpolate text inside single quotes, everything # is printed as is. # \n is for new line and \t is for tab. You can escape (overrule its # special meaning in perl) a character by prefixing it with # backslash \, e.g. \" \\ # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/functions/print.html # https://en.wikipedia.org/wiki/ASCII # ------------------------------------------------------------------ my $human_genes = 20000; print "You have $human_genes genes\n"; print ("You have $human_genes genes\n"); print "You have "; print "$human_genes"; print " genes\n"; print "You have ", $human_genes, " genes\n"; print "You have \"$human_genes\" genes\n"; print 'You have \"$human_genes\" genes\n'; print "\n"; my $bell = chr(7); print $bell;[csc210@boston myPerl]$ ./print_out.pl You have 20000 genes You have 20000 genes You have 20000 genes You have 20000 genes You have "20000" genes You have \"$human_genes\" genes\n
Download

-
BRAC2 mRNA:
Visit the Web page
http://www.ncbi.nlm.nih.gov/nuccore/224004157?report=genbank
containing a partial coding mRNA sequence of the breast cancer gene BRAC2. You can read more about this gene at https://ghr.nlm.nih.gov/gene/BRCA2
Observe that the first 60 nucleotides of this gene are:gggtgcgacg attcattgtt ttcggacaag tggataggca accactaccg gtggattgtc
Create a Perl program that stores this sequence without the blank spaces in a string and prints out the string in lower case and also in upper case letters. - ASCII art:
Write a Perl program that prints out the image below.
__ | \ |\ | /\ |__/ | \| /--\
Note: You can copy the image and paste it into your editor. To print the escape character \ you need to use \\ .Personal challenge: Design your own letters to print out your initials.
- Finding bugs:
Find and exterminate the bugs in the Perl code below
#!/usr/bin/Perl -w use strict my $firt 10_bp = "gggtgcgacg"; my $second_10_bp = "attcattgtt"; $gene = "BRAC2"; print 'The first 20 bp of $gene are"; print "$first_10_bp$second_10_bp\n";
so that it prints out:The first 20 bp of BRAC2 gene are gggtgcgacgattcattgtt
- Pretty prints:
When precise control with the output of your program is needed
(lining up columns, decimal points, etc.), the
printf function of Perl
is your tool. It takes two arguments:
printf "FORMAT", VARIABLES;
FORMAT examples are: %15s field of width 15 for a string; %6d for an integer of width 6; %4.2f for a floating number with 4 digits to the left and 2 digits to the right of the decimal point.Now, using the statement
printf "%15s %10s %10s \n", "Amino acid", "1-letter", "codon";
as a template, write a Perl code that prints out the tableAmino acid 1-letter codon --------------- ---------- ---------- Serine S AGT AGC Cyteine C TGTThe sprintf function takes almost all of the same options of printf, but it returns the formatted string rather than printing it. This functionaly if useful for storing a formatted string in a variable. Please consult the Perl documentation for further information on option on formats.
Data Structures
Scalars
Download
Arrays
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------- # File Name: arrays.pl # # Arrays are lists of scalars. Array names begin with @. Arrays are # defined by listing their contents in parentheses, separated by # commas. The contents of an array are indexed beginning with 0 (not 1). # Negative index looks from the end of array. # To retrieve the elements of an array, replace the @ sign with a $ # sign, and follow that with the index position of the element you want. # (It begins with a dollar sign because array entries are scalars.) # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/perldata.html # https://en.wikipedia.org/wiki/DNA_codon_table # ------------------------------------------------------------------ my @stop_codons = ("TAA", "tAG"); print "Stop codons are @stop_codons \n"; my $first_stop_codon = $stop_codons[0]; print "First stop codon is $first_stop_codon \n"; $stop_codons[1] = "TAG"; print "Second stop codon is $stop_codons[1] \n"; $stop_codons[2] = "TGA"; print "Stop codons are @stop_codons \n"; my $length_of_array = scalar @stop_codons; print "The length of the stop codons array is $length_of_array \n";[csc210@boston myPerl]$ ./arrays.pl Stop codons are TAA tAG First stop codon is TAA Second stop codon is TAG Stop codons are TAA TAG TGA The length of the stop codons array is 3
Download

Hashes
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: hashes.pl # # A hash is a variable type akin to an array that can hold a number # of values. However, instead of indexing values with integers, it # uses a unique name, called a key, for each entry. The name of a # hash begins with a percentage sign. A hash is defined by # comma-separated pairs of key and value, with a "fat arrow" in # between. # If you want to see what keys are in a hash, you can use the keys # function with the name of the hash. This returns a list containing # all of the keys in the hash. The list is not always in the same # order, though. # Code below illustrates some of the basic operations with hashes. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # https://en.wikipedia.org/wiki/Restriction_enzyme # http://perldoc.perl.org/perldata.html # ------------------------------------------------------------------ my %restriction_enzymes = ("EcoRI" => "GAATTC", "AluI" => "AGCT", "NotI" => "GCGGCCGC", "TaqI" => "TCGA" ); print %restriction_enzymes; print "\n"; my @key_list = keys %restriction_enzymes; print "@key_list\n"; # To get a list of values my @value_list = values %restriction_enzymes; print "@value_list\n"; # To fetch any value from a hash by referring to $hashname{key}, # or modify it in place just like any other scalar. my $EcoRI_value = $restriction_enzymes{"EcoRI"}; print "The recognition site of EcoRI is ", $EcoRI_value, "\n"; # To add to an existing hash $restriction_enzymes{"EcoRV"} = "GATATC"; @key_list = keys %restriction_enzymes; print "@key_list\n"; @value_list = values %restriction_enzymes; print "@value_list\n"; # To delete an entry with a given key delete $restriction_enzymes{"EcoRV"}; @key_list = keys %restriction_enzymes; print "@key_list\n"; @value_list = values %restriction_enzymes; print "@value_list\n";[csc210@boston myPerl]$ ./hashes.pl NotIGCGGCCGCAluIAGCTEcoRIGAATTCTaqITCGA NotI AluI EcoRI TaqI GCGGCCGC AGCT GAATTC TCGA The recognition site of EcoRI is GAATTC NotI AluI EcoRV EcoRI TaqI GCGGCCGC AGCT GATATC GAATTC TCGA NotI AluI EcoRI TaqI GCGGCCGC AGCT GAATTC TCGA
Download

- Array of codons for acidic acids: Create an array variable that holds the DNA codons for Glutamic acid. Next, add to this array the DNA codons that code for Aspartic acid. Your new array now should hold all the DNA codons for acidic acids. Print the codons for acidic acids, one per line. You will need to look up the necessary codons in a DNA codon table.
-
Push and pop:
Sometimes it is convenient to add to or subtract from an array without worrying about
indecies.
The
push function adds an element(s) to the end of an array:
push(@codon_array, "GAT");
Now, redo the previous problem using the push function.The pop function of Perl pops and returns the last value of an array, shortening the array by one element:
$last_element = pop(@codon_array);
Analogously, the unshift and shift functions add or remove elements at the start of an array. -
Sorting an array of strings:
Sorting entries of an array is a common task.
The sort function of Perl
takes and array and returns the sorted array:
@sorted = sort @unsorted;
By default, the elements are sorted lexically in standard string comparison order.Modify your Perl code in the previous problem so that your code prints out the codons sorted lexically.
-
Sorting an array of numbers:
The sort function of Perl
can be made to sort according to desired comparisons; you should consults the Perl documentation for further
information.
To sort an array of numbers numerically, use
@unsorted_numbers = (21, 13, 0, -3.1, 21, 33, 2); @sorted_numbers = sort { $a <=> $b } @unsorted_numbers;with the relational comparison operator <=> for numbers and the special variables $a and $b.Now, write a Perl program to sort the unsorted array of numbers above, and print the sorted array. Experiment with or without the block option { $a <=> $b }.
- Hash of codons for acidic acids:
Create a hash variable with keys the RNA codons that code for acidic amino acids,
and values the names of the acidic acids.
Extract the keys array of your hash and print it.
Extract the values array of your hash and print it.
You will need to look up for the necessary codons in a
RNA codon table.
Challenge: For future use, you might consider extending your hash variable for the entire codon table: (key, value) are (codon, amino acid).
String Manipulation
Length
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: length.pl # # length() function takes a string as an argument and returns # the number of characters in the string. # Code below computes the number of nucleotides in a DNA segment. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://www.ncbi.nlm.nih.gov/nuccore/226377833?report=fasta # http://perldoc.perl.org/functions/length.html # http://perldoc.perl.org/index-functions-by-cat.html # ------------------------------------------------------------------ my $zika_DNA = "AGTTGTTGATCTGTGTGAGT"; my $zika_DNA_length = length($zika_DNA); print "The first $zika_DNA_length nucleotides ", "of Zika virus is $zika_DNA \n";[csc210@boston myPerl]$ ./length.pl The first 20 nucleotides of Zika virus is AGTTGTTGATCTGTGTGAGT
Download

Index
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: index_function.pl # # index() function has two forms: # index(string, substring) searches for substring inside string # from the beginning of string; it returns the index (position) of the # first character of substring inside string. If substring is not # found, index returns -1. # index(string, substring, starting_position) searches for substring # inside string from starting_position, not from the beginning. # uc(string) returns string in all uppercase. # lc(string) returns string in all lowercase. # Code below computes the index of start codon in a DNA segment. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://www.ncbi.nlm.nih.gov/nuccore/NC_012532.1?report=genbank # http://perldoc.perl.org/functions/index.html # ------------------------------------------------------------------ my $zika_DNA = "catgtgtgacgccaccatga"; print "Zika DNA segment is $zika_DNA \n"; $zika_DNA = uc($zika_DNA); print "Zika DNA segment is $zika_DNA \n"; my $start_codon = "ATG"; my $first_index = index($zika_DNA, $start_codon); print "First start codon index is $first_index \n"; my $second_index = index($zika_DNA, $start_codon, ($first_index + length($start_codon)) ); print "Second start codon index is $second_index \n"; my $last_index = rindex($zika_DNA, $start_codon); print "Last start codon index is $last_index \n"; # index return -1 if cannot find it. $start_codon = "atg"; my $start_lc = index($zika_DNA, $start_codon); print "Start codon $start_codon index is $start_lc \n";[csc210@boston myPerl]$ ./index_function.pl Zika DNA segment is catgtgtgacgccaccatga Zika DNA segment is CATGTGTGACGCCACCATGA First start codon index is 1 Second start codon index is 16 Last start codon index is 16 Start codon atg index is -1
Download

Substrings
Download

Reverse
Download

Concatenation
Download

Transliteration
Download
Substitution
Download

- Codons in array: Create a Perl program that defines a scalar variable, stores a 15-base long DNA coding sequence in it and then extracts all 5 codons comprising the DNA sequence and stores them into an array. The use of substr() is mandatory. Your program should print the sequence and, on a new line, the codons comprising it, separated by tabs.
- Finding motifs: Create a Perl program that computes and prints the number of nucleotides
that separate the first and last appearance of the motif AGAG in lower case, upper case,
or combination, in the DNA sequence
GACGTCGCCAGAGAggcataTAACGATAtgacacagagagagcaGAGACAAGT
- Reverse complement: Create a Perl program that computes the
reverse complement of the DNA sequence below and prints the sequence and its reverse complement:
GACGTCGCCAGAGAggcataTAACGATAtgacacagagagagcaGAGACAAGT
- Counting base pairs with tr:
The tr operator returns the number of characters that were changed (transliterated).
For example, the Perl statement
$number_of_C = ( $DNA_sequence =~ tr/C/C/ );
captures the numbers of Cs in the string $DNA_sequence and and records it in the variable $number_of_C. Now, write a Perl code that prints out the percentages of the four nucleotides A, C, G, T in the following segment of the breast cancer gene BRAC2:gggtgcgacg attcattgtt ttcggacaag tggataggca accactaccg gtggattgtc
-
Split and join:
In addition to the ones we have explored, there are two additional useful string functions.
The
split
function breaks up a string into substrings according to a given pattern
and stores each substring as an element of an array. For example, the statement
@nucleotides = split( //, $DNA_sequence );
breaks up the $DNA_sequence into individual nucleotides and stores them as an array. Complicated splitting patterns can be specified using "regular expressions." In the example statement, the pattern // splits on nothing.The join function combines all elements of an array into a single string with a specified separator between cells. For example, the statement
$DNA_sequence = join( '', @nucleotides );
will convert the array @nucleotides back to a string. The join is useful in combining lines of a genome or protein file into a single string. Also it can be used to output a list of items separated by, say, tabs, for inclusion in a spread sheet; consult Perl documentation and the references for further information.Now, write a short Perl code that takes the string
my $BRAC2_seq = "gggtgcgacgattcattgttttcggacaag";
prints it; converts it to an array using split and prints the array; reconverts the array back to a string using join and prints the string. Do you get the original string?
Conditionals and Loops
If Else
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: if_else.pl # # if(EXPR){ # # } # else{ # # } # If EXPR is TRUE the first block is executed, otherwise the second # block following else is executed. # Code below determines if the first codon in a DNA segment # is the start codon "ATG" or not and reports the result. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/perlsyn.html#Compound-Statements # ------------------------------------------------------------------ my $DNA_segment = "ATGACATGA"; my $codon1 = substr($DNA_segment, 0, 3); if( ($codon1 eq "ATG") ) { print "Codon $codon1 is a start codon. \n"; } else { print "Codon $codon1 is not a start codon. \n"; } print "Done!\n";[csc210@boston myPerl]$ ./if_else.pl Codon ATG is a start codon. Done!
Download

If Elsif
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: if_elsif.pl # # Code below determines if the last codon in a DNA segment # is the start codon ATG or one of the stop codons TAA, TAG, or TGA; # or none of the above. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/perlsyn.html#Compound-Statements # https://en.wikipedia.org/wiki/DNA_codon_table # ------------------------------------------------------------------ my $DNA_segment = "ATGACATGACCAATC"; my $codon = substr($DNA_segment, -3, 3); if( ($codon eq "ATG") ){ print "Codon $codon is a start codon \n"; } elsif( ($codon eq "TAA") or ($codon eq "TAG") or ($codon eq "TGA") ){ print "Codon $codon is a stop codon \n"; } else{ print "Codon $codon is neither a start nor a stop codon \n"; } print "Done! \n";[csc210@boston myPerl]$ ./if_elsif.pl Codon ATC is neither a start nor a stop codon Done!
Download

While
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: while.pl # # while (EXPR){ # # } # while loop iterates the block as long as EXPR remains TRUE. # Code below first computes the reverse strand of a DNA segment using # Perl's reverse() function. Then, using a while loop develops an # algorithm to reverse a string. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/functions/while.html # http://www.ncbi.nlm.nih.gov/nuccore/226377833?report=fasta # http://www.as.miami.edu/prismrsvp/ # ------------------------------------------------------------------ my $zika_DNA = "CATGTGTGACGCCACCATGA"; print "Original\t $zika_DNA \n"; my $reversed_zika_DNA; $reversed_zika_DNA = reverse($zika_DNA); print "Reversed\t $reversed_zika_DNA \n"; my $length_zika = length($zika_DNA); my $index = $length_zika - 1; my $reversed; while( $index >= 0 ){ $reversed = $reversed.substr($zika_DNA, $index, 1); $index = $index - 1; } print "Ours\t\t $reversed \n";[csc210@boston myPerl]$ ./while.pl Original CATGTGTGACGCCACCATGA Reversed AGTACCACCGCAGTGTGTAC Ours AGTACCACCGCAGTGTGTAC
Download

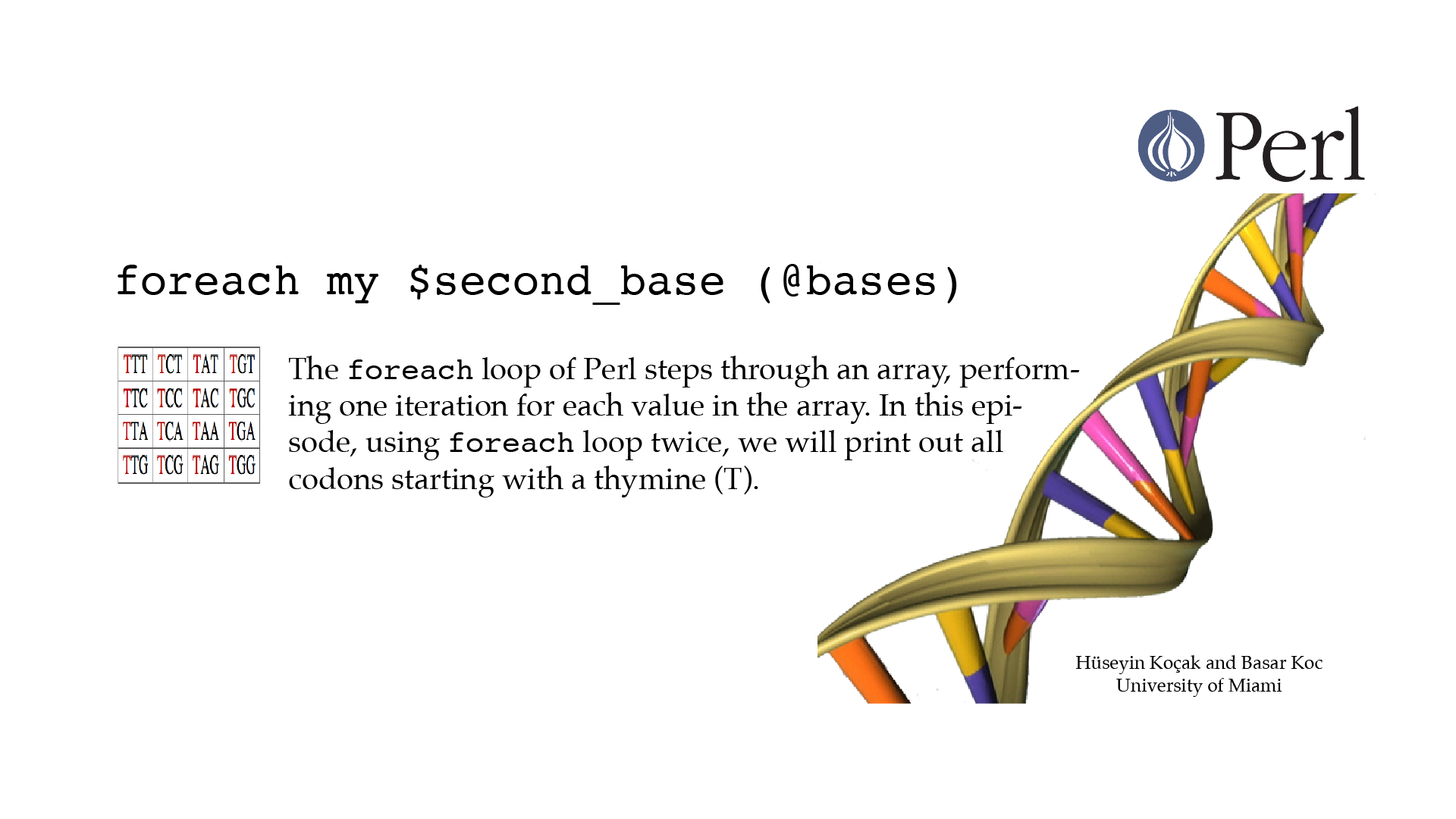
For Each
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: foreach.pl # # The foreach loop steps through an array, performing one iteration # for each value in the array. # Code below prints out all codons starting with T. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # https://en.wikipedia.org/wiki/DNA_codon_table # http://perldoc.perl.org/functions/foreach.html # ------------------------------------------------------------------ my @bases = ("T", "C", "A", "G"); foreach my $base (@bases){ print "$base "; } print "\n"; print "Codons starting with T: \n"; foreach my $second_base (@bases){ print "Codons starting with T$second_base:\n"; foreach my $third_base (@bases){ print "T$second_base$third_base\n"; } } print "\n";[csc210@boston myPerl]$ ./foreach.pl T C A G Codons starting with T: Codons starting with TT: TTT TTC TTA TTG Codons starting with TC: TCT TCC TCA TCG Codons starting with TA: TAT TAC TAA TAG Codons starting with TG: TGT TGC TGA TGG
Download
For
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: for.pl # # Syntax of the for loop for iterations: # for (initialization; test; increment){ # # } # # int() returns the integer part of a decimal number. # # Problem: Assume the population of Florida sandhill cranes grows by # 1.94% annually. If we start with a population of 425 birds, how big # will the population be after 28 years? Using a for loop, code below # computes and prints the population sizes for 28 years. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # https://sora.unm.edu/sites/default/files/journals/jfo/v061n02/p0224-p0231.pdf # https://www.swfwmd.state.fl.us/education/interactive/springscoast/sandhillcranes.shtml # http://perldoc.perl.org/perlsyn.html#Compound-Statements # ------------------------------------------------------------------ # Initial population and growth rate my $population = 425; my $growth_rate = 0.0194; for (my $year = 0; $year <= 28; $year++) { print "At year $year, the population is ", $population, "\n"; # print "At year $year, the population is ", int($population + 0.5),"\n"; $population = $population + $growth_rate * $population; }[csc210@boston myPerl]$ ./for.pl At year 0, the population is 425 At year 1, the population is 433.245 At year 2, the population is 441.649953 At year 3, the population is 450.2179620882 At year 4, the population is 458.952190552711 At year 5, the population is 467.855863049434 At year 6, the population is 476.932266792593 At year 7, the population is 486.184752768369 At year 8, the population is 495.616736972075 At year 9, the population is 505.231701669334 At year 10, the population is 515.033196681719 At year 11, the population is 525.024840697344 At year 12, the population is 535.210322606872 At year 13, the population is 545.593402865446 At year 14, the population is 556.177914881035 At year 15, the population is 566.967766429727 At year 16, the population is 577.966941098464 At year 17, the population is 589.179499755774 At year 18, the population is 600.609582051036 At year 19, the population is 612.261407942826 At year 20, the population is 624.139279256917 At year 21, the population is 636.247581274501 At year 22, the population is 648.590784351227 At year 23, the population is 661.173445567641 At year 24, the population is 674.000210411653 At year 25, the population is 687.075814493639 At year 26, the population is 700.405085294815 At year 27, the population is 713.992943949535 At year 28, the population is 727.844407062156
Download

Count with Hashes
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: count_with_hashes.pl # # Code below counts the number of each codon that appears in a DNA # segment. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/perldata.html # http://www.ncbi.nlm.nih.gov/nuccore/224004157?report=genbank # ------------------------------------------------------------------ my %codon_count; my $DNA_seq = "gggtgcgacgattcattgttttcggacaagtggataggcaaccactaccggtggattgtc"; print "Sequence: $DNA_seq\n"; my $DNA_length = length($DNA_seq); for( my $index = 0; $index <= $DNA_length - 3; $index = $index + 3){ my $codon = substr($DNA_seq, $index, 3); if(not exists $codon_count{$codon}){ $codon_count{$codon} = 0; } $codon_count{$codon} = $codon_count{$codon} + 1; } my @key = keys %codon_count; my @value = values %codon_count; for (my $i=0; $i < scalar(@key); $i++){ print "$key[$i] => $value[$i] \n"; }[csc210@boston myPerl]$ ./count_with_hashes.pl Sequence: gggtgcgacgattcattgttttcggacaagtggataggcaaccactaccggtggattgtc ggc => 1 ggg => 1 att => 1 tgg => 2 ttt => 1 gat => 1 tcg => 1 gtc => 1 aac => 1 aag => 1 tca => 1 ttg => 1 tac => 1 cgg => 1 tgc => 1 ata => 1 gac => 2 cac => 1
Download

- Searching for patterns:
Create a Perl program that can check whether a given DNA sequence
contains the pattern
TATAxxxATGxxxT
where xxx can be any three consecutive DNA bases. That means your program, given some string, should identify if TATA, followed by any three DNA base pairs, followed by ATG, followed by any three DNA base pairs, followed by T can be found in the given string. Check both the forward and reverse complementary strand (reverse complement). Create three examples, one with a DNA string that contains the pattern in the forward strand, one that contains the pattern in the reverse complement strand and one that does not contain it in either strand, to demonstrate how the program works. Please include print statements that identify each different case and the position of the pattern in the string, if found. - Codons in array: Create a Perl program that stores a DNA sequence of any length, extracts all codons in the sequence, and stores them in an array. Print the sequence and the codons, one per line, comprising it. Your program should handle the case where the sequence length is not a multiple of 3. Test your program with several sequences.
- Logistic growth with while and for:
Consider the discrete logistic model
x[n+1] = r * x[n] * (1.0 - x[n])
governing the growth of a single population with non-overlapping generations. Here, r is the growth rate and x[n] is the density of the n-th generation. Fix the growth rate and the initial population density asr = 3.73 and x[0] = 0.43.
Write a Perl program that computes the population densities for 12 generations using a while statement, and prints out each generation and its population density on a separate line for all 12 generations.Now, write a second Perl program that accomplishes the same task using a for statement.
- Locating cutting sites of restriction enzymes:
In the episode on hashes, we created a hash varible with the names of restriction enzymes as keys
and the corresponding recognition sequences as values:
my %restriction_enzymes = ("EcoRI" => "GAATTC", "AluI" => "AGCT", "NotI" => "GCGGCCGC", "TaqI" => "TCGA");Write a Perl code to determine if the DNA sequencemy $DNAseq = "GGCGATGCTAGTCGCGTAGTCTAAGCTGTCGAGAATTCGGATGTCATGA";
will be cut with any of these restriction enzymes. Print out the location (index) of the recognition sites in the DNA sequence. For this problem, you should loop through the keys of the hash variable. - Generating random DNA sequences: Generate a random DNA string 101 bases
long, store it in a string variable, and print it. To do that, use an
array of nucleotides, and the
rand
Perl function, to generate random numbers. Other tools that you can use include:
string concatenation, the
int Perl function,
and a loop.
Run your program several times. Do you get the same random sequence? Unlikely! For certain simulations, it may be necessary to use the same random sequence. Consult the documentation on how to set the seed of the srand Perl function.
- Generating random DNA sequences with a specified frequency: Chargaff rule states that "a double-stranded DNA molecule globally has percentage base pair equality: %A = %T and %G = %C." However, %A and %G varies with organisms. For instance, humans have, approximately, 29% A and 21% C. Write a Perl program that generates random DNA sequences with percentages of A, T, C, and G of human DNA.
- All codons: Using as many foreach as necessary, write a Perl program that prints out all 64 codons.
Scope and Subroutines
Scope
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: scope.pl # # With use strict; the scope of a variable is from the point of # declaretion to end of the block it is declared in. # Code below illustrates concept of scope of variables with a naked # block { } and a for loop. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/strict.html # ------------------------------------------------------------------ my $RNA = "AGU"; my $length_RNA; $length_RNA = length($RNA); print "Before block: length = $length_RNA \n"; { my $RNA = "UGGAUG"; $length_RNA = length($RNA); print "Inside block: length = $length_RNA \n"; } $length_RNA = length($RNA); print "After block: length = $length_RNA \n"; print "\n"; my $i = 200; print "Before for loop: i = $i\n"; for(my $i = 0; $i < 6; $i++){ print "Inside for loop: i = $i\n"; } print "After for loop: i = $i\n";[csc210@boston myPerl]$ ./scope.pl Before block: length = 3 Inside block: length = 6 After block: length = 3 Before for loop: i = 200 Inside for loop: i = 0 Inside for loop: i = 1 Inside for loop: i = 2 Inside for loop: i = 3 Inside for loop: i = 4 Inside for loop: i = 5 After for loop: i = 200
Download

Subroutines
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: subroutines.pl # # Arguments (parameters) passed to a subroutine are automatically # stored in a special array named @_ which is private to the # subroutine. For example, statement my ($arg1, $arg2) = @_ # creates two private variables and sets their values. # Subroutine below takes a DNA segment and returns the corresponding # RNA segment as a string. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://www.ncbi.nlm.nih.gov/nuccore/226377833?report=fasta # http://perldoc.perl.org/functions/sub.html # ------------------------------------------------------------------ my $zika_DNA_segment = "AGTTGTTGATCTGTGTGAGTCAGACTGCG"; print "Zika DNA segment is $zika_DNA_segment. \n"; my $zika_RNA_segment; $zika_RNA_segment = DNAtoRNA($zika_DNA_segment); print "Zika RNA segment is $zika_RNA_segment. \n"; # *********************************************** # DNAtoRNA # *********************************************** # This subroutine takes a DNA sequence and returns # corresponding RNA sequence. # In : DNA sequence as a string scalar variable. # Out : RNA sequence as a string scalar variable. # *********************************************** sub DNAtoRNA{ my ($segment) = @_; $segment =~ tr/Tt/Uu/; return $segment; }[csc210@boston myPerl]$ ./subroutines.pl Zika DNA segment is AGTTGTTGATCTGTGTGAGTCAGACTGCG. Zika RNA segment is AGUUGUUGAUCUGUGUGAGUCAGACUGCG.
Download

- Change the scope:
The Perl code below does not run:
#!/usr/bin/perl -w use strict; for(my $i = 0; $i < 6; $i++){ print "Inside for loop: i = $i\n"; } print "After for loop: i = $i\n"Modify the code above so that it prints outInside for loop: i = 0 Inside for loop: i = 1 Inside for loop: i = 2 Inside for loop: i = 3 Inside for loop: i = 4 Inside for loop: i = 5 After for loop: i = 6
- A subroutine returning a random sequence: Write a Perl subroutine that takes as a parameter the length and returns a random DNA sequence with the requested length. Make certain that the variables are local to your subroutine.
- A subroutine for growth of Florida sandhill cranes:
In the while episode, we encountered the following problem:
The population of Florida sandhill cranes in the Okefenokee swamp,
under the current environmental
conditions, grows by 1.94% annually. If we start with a population of
425 birds, how big will the population be after 28 years?
Now, create a Perl subroutine that, given an initial
population and a number of years, can calculate and return the Florida sandhill
crane population after that time period. Test your subroutine
with several examples to verify it works properly.
Now modify the subroutine to accept the growth rate as a parameter as well. Test your new code with different growth rates.
Keyboard
Stdin
is used to get a scalar value from # the keyboard into a Perl program. # Code below captures a line entered from the keyboard, removes the # new line character "\n", and echoes the line to the screen. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/perlvar.html # http://perldoc.perl.org/functions/chomp.html # ------------------------------------------------------------------ my $keyboard_entry; print "Please type something and ENTER: "; $keyboard_entry = <STDIN>; # get the keyboard entry chomp($keyboard_entry); # remove newline character from line print "You typed: $keyboard_entry \n"; [csc210@boston myPerl]$ ./stdin.pl Please type something and ENTER: Perl for Biologists You typed: Perl for Biologists
Download

Argv
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: argv.pl # # Command line arguments are provided directly after the name of # the program. The command line arguments are stored in an array # called @ARGV. # $#ARGV is the length of ARGV - 1. # Our program will print out the number and the list of arguments; # will exit if no argument is provided. # Example usage: ./args.pl red blue # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/perlvar.html # ------------------------------------------------------------------ if($#ARGV < 0) { die "Please provide a command line argument!\n"; } print "First argument is $ARGV[0] \n"; print "Total number of arguments is ", scalar(@ARGV)," \n"; foreach my $argument (@ARGV){ print "$argument\n"; }[csc210@boston myPerl]$ ./argv.pl Please provide a command line argument! [csc210@boston myPerl]$ ./argv.pl Perl Biology First argument is Perl Total number of arguments is 2 Perl Biology
Download

- Keyboard inputs: Write a Perl program that asks the user to enter a DNA sequence from the keyboard and captures the sequence in a variable. Next, your program should ask the user for a second shorter DNA sequence to be entered. Now, your program should report if the second sequence is a substring of the first string.
- Inputs as an array:
In array context, the line-input operator <STDIN> returns each input line as an element of an array:
my @input_lines = <STDIN>;
If the input is from a file, all the lines in the file are read. If the input is from the keyboard, you need to signal the end of input by typing CTRL-D in Linux or OSX, and CTRL-Z in Windows.Write a Perl program that reads a list of strings, one per line, and prints out the strings in lexically sorted order.
First, test your program with several lines of input from the keyboard. Next, put your input lines in a file, say input_strings.txt. Now use the Unix redirection operator < to give your file as input to your program:
./my_program.pl < input_strings.txt
- Arguments as input: Write a Perl program that takes two strings as command-line arguments and reports if the second argument is a substring of the first argument.
Files
Write to File
#!/usr/bin/perl -w use strict; # ----------------------------------------------------------------- # File name: write_to_file.pl # # Code below opens a file and writes information about a DNA sequence, # in FASTA format. Finally, it reports if the file is created. # You can change the filename to a file of your choosing. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://www.esa.int/Our_Activities/Space_Science/Rosetta/Rosetta_s_comet_contains_i$ # http://https://en.wikipedia.org/wiki/DNA_codon_table # http://perldoc.perl.org/functions/-X.html # http://perldoc.perl.org/functions/open.html # ---------------------------------------------------------------- my $filename = "Rosetta_partial.fasta"; open (FILE_TO_WRITE, ">", "$filename") # ">" to overwrite or die "Cannot open $filename to write: $!"; # ">>" to append print FILE_TO_WRITE ">gi|210|ref|CSC_210.1| Rosetta partial genome\n"; print FILE_TO_WRITE "ATG\n"; print FILE_TO_WRITE "GGT"; print FILE_TO_WRITE "GGC"; print FILE_TO_WRITE "GGA"; print FILE_TO_WRITE "GGG\n"; print FILE_TO_WRITE "TGA"; close(FILE_TO_WRITE); if (-e $filename){ print "Rosetta partial genome is written to $filename file successfully!\n"; }[csc210@boston myPerl]$ ./write_to_file.pl Rosetta partial genome is written to Rosetta_partial.fasta file successfully!
Download

Read from File
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------- # File name: read_from_file.pl # # This program opens a file, then reads the file line by line and # prints out the lines to screen. # Program exits if the file does not exist or cannot be opened. # Currently, the file to read is the file containing this code. # You can change the filename to a file of you choosing. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/functions/open.html # ------------------------------------------------------------------ my $filename = "read_from_file.pl"; open (FILE_TO_READ, "<", "$filename") or die "Cannot open file $filename : $!"; while (my $line = <FILE_TO_READ>) { print "$line"; } close (FILE_TO_READ);[csc210@boston myPerl]$ ./read_from_file.pl #!/usr/bin/perl -w use strict; # ------------------------------------------------------------------- # File name: read_from_file.pl # # This program opens a file, then reads the file line by line and # prints out the lines to screen. # Program exits if the file does not exist or cannot be opened. # Currently, the file to read is the file containing this code. # You can change the filename to a file of you choosing. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/functions/open.html # ------------------------------------------------------------------ my $filename = "read_from_file.pl"; open (FILE_TO_READ, "<", "$filename") or die "Cannot open file $filename : $!"; while (my $line = <FILE_TO_READ>) { print "$line"; } close (FILE_TO_READ);
Download

Get from NIH
#!/usr/bin/perl -w use strict; use LWP::Simple; # ------------------------------------------------------------------- # File name: get_fasta_nih.pl # # This program downloads a fasta file from NCBI. # GeneId or accession number and the name of the local file to save in # must be provided as command line arguments. # Example usage: ./get_fasta_nih.pl XM_002295694.1 BRCA2.fasta # You can change the filename to a file of you choosing. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://www.ncbi.nlm.nih.gov/nuccore/XM_002295694.1?report=fasta # http://www.ncbi.nlm.nih.gov/books/NBK25501/ # http://perldoc.perl.org/perlfaq9.html # ------------------------------------------------------------------ if ($#ARGV < 1) { die "Please supply a GeneId(gi) number and a file name as arguments!"; } my $gi = $ARGV[0]; my $filename = $ARGV[1]; my $url = "http://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi"; my $db = "db=nuccore"; my $id = "id=$gi"; my $rettype = "rettype=fasta"; my $retmode = "retmode=text"; getstore("$url?$db&$id&$rettype&$retmode", $filename);[csc210@boston myPerl]$ ./get_fasta_nih.pl Please supply a GeneId(gi) number and a file name as arguments! at ./get_fasta_nih.pl line 23. [csc210@boston myPerl]$ ./get_fasta_nih.pl XM_002295694.1 BRCA2.fasta
Download

Read Fasta File
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------- # File name: read_fasta_file.pl # # We will open a file containing information about a DNA sequence, # in FASTA format. Then we will read the file line-by-line, except # line starting with > sign; remove newline characters and concatenate # lines into a single string. # The filename will be provided as a command line argument. # This means that it will be supplied when we run our program, # directly after the name of the program. # Example: ./read_fasta_file.pl BRAC2.fasta # The command line arguments are stored in an array called @ARGV. # $#ARGV is length of @ARGV minus 1. # Our program will exit if the number of arguments is not 1, # or the file does not exist or cannot be opened. # You can change the filename to a file of your choosing. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # https://en.wikipedia.org/wiki/BRCA2 # http://www.ncbi.nlm.nih.gov/nuccore/224004157?report=fasta # ------------------------------------------------------------------ if ($#ARGV != 0) { die "Please supply a file name as a command line argument!"; } my $filename = $ARGV[0]; open (FILE_TO_READ, "<", "$filename") or die "Cannot open file $filename : $!"; my $sequence; while (my $line = <FILE_TO_READ>) { if ($line !~ /^>/) { chomp($line); $sequence = $sequence.$line; } } print "$sequence\n"; close (FILE_TO_READ);[csc210@boston myPerl]$ ./read_fasta_file.pl Please supply a file name as a command line argument! at ./read_fasta_file.pl line 29. [csc210@boston myPerl]$ ./read_fasta_file.pl BRAC2.fasta GGGTGCGACGATTCATTGTTTTCGGACAAGTGGATAGGCAACCACTACCGGTGGATTGTCTGGAAGCTAGCAGCAATGGAGAGACGGTTTCCACACCATCTTGGAGGACATTACTTGACGTACGAGCGTGTGCTGAAACAAATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAGACGGCCTGCAGTACGCATAATGCTCAACCGAGATGTTGCAGCGAGTTTGCCAGTCATCTTATGCGTAAGCCAAATCCTTCGATTCAAATCAAGACCGCCAAAAGGAAGTTCTTCCGACGAGATCAAAGAAGAAGTCCGACTGGAGTTGACGGATGGATGGTACTCACTACCTGCTGTAGTGGACGAAATACTGTTGAAGTTTGTTGAAGAAAGGAGAATCGCAGTGGGATCAAAACTAATGATTTGCAATGGGCAGTTAGTTGGATCTGATGACGGAGTGGAGCCTCTCGATGACAGCTACTCATCTTCCAAACGAGATTGTCCTCTATTGCTGGGCATCTCTGCCAACAACTCCCGTTTAGCAAGATGGGATGCAACTCTAGGTTTTGTACCTCGCAACAACTCTAATCTATACGGCGGCAATCTTTTGGTCAAATCCCTGCAAGACATTTTCATCGGCGGAGGTACTGTTCCGGCTATTGATTTGGTTGTTTGTAAGAAGTACCCAAGGATGTTTCTAGAGCAATTAAACGGTGGAGCTTCCATTCATCTTACAGAAGCCGAAGAAGCAGCACGCCAAAGTGAGTACGATTCAAGGCATCAGCGAGCAAGCGAGAGATATGCCGACGATGCTACGAAGGAATGTTCAGAGGTAAGTTCATTGCTGTTCACATTCTTCACTATGAAGCCACTTCCGTTGCTTTGGTACAATCTTGTCACTGACTCATCTTTTGGCGTTCATGATTCGCACAGGAAATCGATGAGGATGCTCCTACTCAGTGGAAAGAGATG
Download

-
Counting Zika virus genome bps:
Download the FASTA file (NC_012532.1) containing the
Zika virus genome. (You can use the "Send" widget on the upper-right corner of
the NCBI Web page containing the genome to download a FASTA file.)
Now, write a Perl program that reads the file, stores the sequence without white
characters (spaces, end-of-line, etc.),
and prints out the number of nucleotides (bps) in the complete Zika virus genome.
- Start codons in the Zika virus:
Modify your Perl code in the previous problem so that your code
prints out the locations of the first appearances of the start codon ATG and one
of the stop codons (TAA, TAG, TGA) afterwords in the Zika genome.
- Logistic growth model:
Consider the discrete logistic model
x[n+1] = r * x[n] * (1.0 - x[n])
governing the growth of a single population with non-overlapping generations. Here, r is the growth rate and x[n] is the density of the n-th generation. Fix the growth rate r = 3.1 and initial population density x[0] = 0.43. Write a Perl program that computes the population densities for 12 generations and writes them out to a file, each generation and its population density on a separate line, as below:At year 0, the population density is 0.43 At year 1, the population density is ...
-
Comparing k-mers in viral genomes:
Create a Perl program to do the following:
- Open a FASTA file whose name is provided as a command line argument, concatenate the sequence lines in a string.
- Extract all substrings of length 9 (9-mers) from the sequence and store all 9-mers in a hash, together with the number of times they appear in the string. Use the 9-mers as keys and the number of appearances as values in the hash. You can check whether a 9-mer has already been inserted in the hash with the 'defined' function. At the end, the program should print all 9-mers and their counts.
- Now, edit the previous program (or create a new one) that opens and processes two separate virus genomes in FASTA format. Your goal is to compare the two genomes and determine the number of substrings of length 9 (9-mers) that they share. Please print all 9-mers that the two genomes share and their total number (count). Select two random genomes, preferably not longer than 10000 nucleotides each. You should supply the FASTA files with the virus genome sequences as command-line arguments to your program and use the @ARGV array to import the sequences.
- The two virus genomes can be downloaded from NCBI. For a starting point, you can use this webpage. Please include your choices virus names and accession numbers in your deliverable.
- Would you expect to get similar results if these were not virus genome sequences but random DNA/RNA sequences?
Regular Expressions
Motif Match
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: motif_match.pl # # Match operator m// looks for PATTERN in STRING; the statement # STRING =~ m/PATTERN/ becomes TRUE if PATTERN is found. PATTERN is # specified using regular expressions. # This program reports if a motif (substring with a specific pattern) # is present in a DNA sequence. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/functions/m.html # http://www.ncbi.nlm.nih.gov/nuccore/224004157?report=fasta # ------------------------------------------------------------------ my $DNA_sequence = "AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG"; my $motif = "ATG.*TAA"; if ($DNA_sequence =~ m/$motif/){ print "Found the motif \n"; } else{ print "Could not find the motif \n"; }[csc210@boston myPerl]$ ./motif_match.pl Found the motif
Download

Motif Match Memory
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: motif_match_memory.pl # # Match operator m/()/ with memory searches for PATTERN in STRING; # the statement STRING =~ m/(PATTERN)/ becomes TRUE if PATTERN is # found; moreover, the found pattern is saved in a special variable # $1. PATTERN is specified using regular expressions. # This program reports if a motif (substring with a specific pattern) # is present in a DNA sequence. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/functions/m.html # http://www.ncbi.nlm.nih.gov/nuccore/224004157?report=fasta # ------------------------------------------------------------------ my $DNA_sequence = "AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG"; my $motif = "ATG.*?TAA"; $DNA_sequence =~ m/($motif)/; if(defined $1) { print "Found the motif $1 \n"; } else { print "Could not find the motif \n"; }[csc210@boston myPerl]$ ./motif_match_memory.pl Found the motif ATGAAGGGCCGCTACGATAA
Download

Motif Match Interactive
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: motif_match_interactive.pl # # Match operator m/()/ with memory searches for PATTERN in STRING; # the statement STRING =~ m/(PATTERN)/ becomes TRUE if PATTERN is # found; moreover, the found pattern is saved in a special variable # $1. PATTERN is specified using regular expressions. # Code below saves a sequence in string and asks the user for a motif. # Program reports if the motif is found in the string. # If a blank line in entered, the program exits. # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/perlre.html#Regular-Expressions # http://perldoc.perl.org/functions/eval.html # http://perldoc.perl.org/functions/qr.html # http://www.ncbi.nlm.nih.gov/nuccore/224004157?report=fasta # Example motifs to try: ATG ^A.*G ^A.*?G [AT]ATT # * T[AG][AG] AT{2,}G AT{2,}C # ------------------------------------------------------------------ my $sequence = "AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG"; print "Sequence = $sequence\n"; # In a loop, ask the user for a motif, search for the motif, # and report if it was found. # Exit if no motif is entered. my $motif; while( ) #An infinite loop { print "Enter a motif to search for: "; $motif = <STDIN>; if( $motif =~ m/^\s*$/ ){ # to check for blank line last; # to leave the while loop } chomp($motif); # Remove the newline at the end of $motif # Check if the motif is a legal regular expression eval{ qr/$motif/ }; if( $@ ){ print "Motif $motif is an illegal regular expression! \n"; } else{ # Look for the motif and save it if found. # found motif is saved in special variable $1 $sequence =~ m/($motif)/; if( defined $1 ){ print "I found $1 \n"; } else{ print "I could not find motif $motif \n"; } } } #end of while loop print "Bye!\n";[csc210@boston myPerl]$ ./motif_match_interactive.pl Sequence = AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG Enter a motif to search for: ATG I found ATG Enter a motif to search for: ^A.*G I found AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG Enter a motif to search for: ^A.*?G I found AATG Enter a motif to search for: [AT]ATT I found AATT Enter a motif to search for: Bye!
Download

Motif Match Global
#!/usr/bin/perl -w use strict; # ------------------------------------------------------------------ # File name: motif_match_global.pl # # Match operator m/()/g with memory searches for PATTERN in STRING # globally; the statement STRING =~ m/(PATTERN)/g becomes TRUE if # PATTERN is found; moreover, the found pattern is saved in a special # variable $1. PATTERN is specified using regular expressions. # This program finds all the occurrences of a motif # in a DNA sequence and reports the motifs found, their locations, # and the total number of found motifs. # pos() function returns the location just after the match # the motif in the regular expression (here consists of # substrings of A and/or T of lengths between 3 and 6). # # Version: 2.1 # Authors: H. Kocak and B. Koc, University of Miami # References: # http://perldoc.perl.org/functions/m.html # http://www.ncbi.nlm.nih.gov/nuccore/224004157?report=fasta # ------------------------------------------------------------------ my $sequence = "AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG"; print "Sequence = $sequence \n"; my $motif = "[AT]{3,6}"; my $matched_motif; #will hold the captured match my $matched_location; #will hold the index of the match my $number_of_matches = 0; #will hold the number of matches while( $sequence =~ m/($motif)/g ) #g is for global, all matches { $matched_motif = $1; $matched_location = pos($sequence) - length($matched_motif); $number_of_matches = $number_of_matches + 1; print "Match $number_of_matches: $matched_motif at $matched_location\n"; } print "\nTotal number of matches is: $number_of_matches\n";[csc210@boston myPerl]$ ./motif_match_global.pl Sequence = AATGAAGGGCCGCTACGATAAGGAACTTCGTAATTTCAG Match 1: AAT at 0 Match 2: ATAA at 17 Match 3: TAATTT at 30 Total number of matches is: 3
Download

-
Deciphering regular expressions:
Give an English translation and an example of each of the following regular expressions:
AT+C [AT]+ [^AT]+ ((.)(.).*\2\3.*\2\3) ^ACTA{3,}TTT$Hint: You can test your examples with the program in motif_match_interactive episode with appropriate sequences.
-
What does it do?
What does the Perl code below do?
#!/usr/bin/perl -w use strict; my $DNA_seq = "AGTTGTTGATCTGTGTGAGTCAGACTGCGACAGTTCGAGTCTGAAGCGAGAGCTAACAACAGTATCAACA"; my $magic_number = 10; while ( $DNA_seq =~ m/(.{1,$magic_number})/g ) { print "$1 \n"; } -
Make it do! Modify the Perl code in the previous problem
so that the code prints out the following:
A GT TGT TGAT CTGTG TGAGTC AGACTGC GACAGTTC GAGTCTGAA GCGAGAGCTA ACAACAGTATC AACA
-
Searching for patterns:
Create a Perl program that can check whether a given DNA sequence
contains the pattern
TATAxxxATGxxxT
where xxx can be any three consecutive DNA bases. That means your program, given some string, should identify if TATA, followed by any three DNA base pairs, followed by ATG, followed by any three DNA base pairs, followed by T can be found in the given string. Check both the forward and reverse complementary strand (reverse complement). Create three examples, one with a DNA string that contains the pattern in the forward strand, one that contains the pattern in the reverse complement strand and one that does not contain it in either strand, to demonstrate how the program works. Please include print statements that identify each different case and the position of the pattern in the string, if found. -
Who is right?
Write a Perl program that can print the number of times a substring with five
characters that starts with 'AT' and ends with 'A' appears in a given string.
This means that you should recognize the appearance of every pattern 'ATxxA',
where 'x' can be any character. Test your program with the following string:
ATAGACATATAGATATGTGCACATAGAGAGATATACACATATATGTACATACA
- Write the program using regular expressions (without using substr).
- Write the program using substr (and no regular expressions).
- The actual number of times that the aforementioned pattern appears in the string above is 9. Was this number calculated correctly by both of your programs? If not, why not?
-
Binding sites in mRNA:
Shine-Dalgarno sequence is a binding site in the mRNA, usually located 8
base pairs upstream of the start codon AUG. The concensus sequence is AGGAGG. Create
a program that can examine any given FASTA sequence for occurences of
the Shine-Dalgarno sequence upstream of a start codon.
You will have to examine both the forward and complementary strands.
You have to report
the occurence of the AGGAGG pattern before any ATG, with 0-20
bases separating them.
Report separately the number of occurences for each length value of the segment between the two sequences. For example, you should report the number of times AGGAGG appears directly before ATG, the number of times AGGAGG appears one base before ATG, etc., up to 20 bases between them.
Now test your code with the genomes of two bacterial chromosomes, both larger than 5MB, one from a gram-negative bacterium and another from a gram-positive bacterium. For a gram negative, you could download the genome of Pseudomonas Aeruginosa, where for gram positive you could use Desulfitobacterium hafniense, or select other genomes of your choice.
- Logistic growth model:
Consider the discrete logistic model
x[n+1] = r * x[n] * (1 - x[n])
modeling the growth of a single population, where x[n] is the density of the population at n-th generation and r is the growth rate. Fix the growth rate r = 3.1 and initial population density x[0] = 0.43. Write a Perl program that computes the population densities for 20 generations and puts them in an array. Then, print out each generation and its population density on a separate line for all 20 generations. - TATA-Pribnow box: Motifs TATAAA or TATAAT of six nucleotides, known as TATA boxes, are an essential part of a promoter site on DNA for transcription to occur. Write a Perl program that finds all the TATA boxes in a DNA sequence and reports their types and locations. If there is no such a box, your program should report that as well. Test your program on several DNA sequences with none, and with several TATA boxes of both types.
- TATA boxes in ebola virus genome: Download the FASTA file (accession number KC545393) containing the ebola virus genome. (You can use the "Send" widget on the upper-right corner of the NCBI Web page containing the genome to download a FASTA file.) Now, modify your program from the previous problem to produce the same TATA box report of the ebola virus genome.
-
Palindromic restriction sites in Zika virus genome:
Restriction enzymes are enzymes that cut double-stranded DNA at specific recognition nucleotide sequences known as
restriction sites. Quite often, these sites are palindromic, which for double-stranded DNA means they read the same
on the reverse strand as they do on the forward strand when both are read in the same orientation. For example, the
following is a palindromic restriction site:
5'-GTATAC-3' |||||| 3'-CATATG-5'
Create a program that, given a DNA sequence, will output all palindromic DNA sites of length 6 and their location. Test your program with:
- The sequence: "GCTAGTGTATGCATGAGCGTAGGCGA TGTGGCGCCGAGCTGAGGTGATCACGTGATGTGCTAGTCG".
- Zika virus genome: Download the FASTA file (NC_012532.1) containing the Zika virus genome. (You can use the "Send" widget on the upper-right corner of the NCBI Web page containing the genome to download a FASTA file.)
| Special characters | Meaning |
|---|---|
| . | any character, except newline |
| \n | newline |
| \t | tab |
| \s | any whitespace character (space, newline, tab) |
| \S | any non-whitespace character |
| \d | any digit character |
| \D | any non-digit character |
| \w | any one word character (alphanumeric plus _) |
| \W | any one nonword character |
| * | match 0 or more times preceding character or group |
| + | match 1 or more times preceding character or group |
| ? | match 0 or 1 time |
| ? | shortest (non-greedy) match |
| { } | repetition |
| { , } | repetition, min to max |
| ( ) | capture |
| ( ) | grouping |
| \1 | recall the first capture |
| \2 | recall the second capture |
| \n | recall the n-th capture |
| ^ | beginning of string |
| $ | end of string |
| [ ] | any one of set of characters |
| [^ ] | except any one of set of characters |
| | | alternative |
| \ | special or quote |
| (?=...) | Positive look-ahead. Matches if ... matches next, but doesn’t consume any of the string |
| (?!...) | Negative look-ahead. Matches if ... doesn’t match next |
| Regular Expression | Example | Meaning |
|---|---|---|
| AGA | TAGATC | match AGA anywhere |
| ^AGA | AGATGC | AGA at the beginning |
| TAA$ | AAGTAA | TAA at the end |
| A.T | AAACTG | A followed by any single character (except newline), followed by T |
| A.*T | CATATCT | A followed by any number of characters, followed by T (greedy) |
| A.*?T | CATATCT | A followed by any number of characters, followed by T (non-greedy) |
| (A.*?T) | CATATCT | capture A followed by any number of characters, followed by T (non-greedy) |
| A{5} | TAAAAATC | 5 A's |
| TA{2,4}CG | CTAAACGA | T followed by 2 to 4 A's, followed by CG |
| [AT]CG | CCTTCGA | either of ACG or TCG |
| [AA|CC|TT]CG | ACCCGTA | AA or CC or TT, followed by CG |
| A(CG){3}T | GACGCGCGTA | A followed by 3 CG's, followed by T |
| ((.)(.)\3\2) | GAATTAC | capture 4 consecutive characters, 1st and 4th, and 2nd and 3rd the same |
| A\.T | TCGA.TAA | A followed by . and T |
| AAA(?=TAG|TGA|TAA) | TAAATGAT | AAA if followed by TAG or TGA or TAA |
- Useful links:
- The Perl Programming Language
This is the official Web site for Perl. You can download Perl for Windows; Unix/Linux and Mac OS X include Perl. - Perl functions and their arguments
- Regular expressions summary with examples
- perlrequick - Perl regular expressions quick start
- Perl for Exploring DNA, by Mark D. LeBlanc and Betsey Dexter Dyer, Oxford University Press, 2007.
- Beginning Perl for Bioinformatics, by James Tisdall, O'Reilly Press, 2001.
- Learning Perl, by Randal L. Schwartz, Tom Phoenix, and Brian D. Fox, O'Reilly Press, 2005.
- NCBI Genome site
- NCBI Zika virus, complete genome (NC_012532.1)
- NCBI Bundibugyo ebolavirus isolate EboBund-112 2012, complete genome (KC545393.1)
- NCBI Thalassiosira pseudonana CCMP1335 chromosome 7 breast cancer 2 early onset (BRAC2) mRNA, partial cds (XM_002295694.1)
- Pan troglodytes verus isolate MABEL mitochondrial D-loop (Chimp (AF176731.1)
- H.sapiens mitochondrial DNA for D-loop (Human) (X90314.1) and
- The Entertainer, by Scott Joplin.